
L’Agenzia Europea per i Medicinali (EMA) ha annunciato nell’ottobre 2024 l’adozione delle attese linee guida sulla Qualità e l’Equivalenza dei Prodotti Topici. Il documento finale è stato redatto nel 2018 ed entrerà in vigore nell’aprile 2025 con il titolo ufficiale ” Linee guida sulla qualità e l’equivalenza dei prodotti cutanei ad azione locale “.
I nostri dipartimenti di IVRT e IVPT e di Garanzia della Qualità hanno riassunto gli aspetti più rilevanti delle linee guida EMA in materia di IVRT e IVPT, suddividendoli tra le raccomandazioni sulla qualità e la sezione sull'equivalenza terapeutica. Infine, hanno condiviso l'esperienza di Kymos Group nel lavorare in conformità alle linee guida sin dalla loro precedente bozza del 2018.
Qual è la nuova linea guida dell’EMA sui prodotti topici per il 2024?
Come suggerisce il titolo, questa linea guida si riferisce ai medicinali ad applicazione locale e ad azione locale per uso cutaneo, ma può essere rilevante anche per altri prodotti, come i preparati per uso auricolare o oculare e i prodotti vaginali ad azione locale. Questo nuovo regolamento è suddiviso in raccomandazioni sulla qualità e sull’equivalenza e introduce un approccio più strutturato alla valutazione dei prodotti cutanei.
La seconda parte relativa all’equivalenza non si applica ai medicinali biologici, ai medicinali a base di erbe, ai prodotti la cui equivalenza in termini di efficacia è dimostrata da studi clinici e ai prodotti in cui la forma farmaceutica del test e quella di riferimento non sono le stesse. Questa esclusione è particolarmente importante quando si tratta di forme farmaceutiche ad assorbimento sistemico come i cerotti transdermici.
Con questi recenti aggiornamenti, l’EMA sottolinea un approccio più strutturato e graduale che dovrebbe semplificare il processo di dimostrazione dell’equivalenza terapeutica, concentrandosi sui metodi in vitro (test di rilascio in vitro o IVRT e test di permeazione in vitro o IVPT) e farmacocinetici (PK) che presentano alternative economiche ed efficienti in termini di tempo agli studi clinici.
Raccomandazioni sulla qualità dei prodotti topici
Le raccomandazioni sulla qualità contenute nelle linee guida si applicano alle nuove domande di autorizzazione all’immissione in commercio e alle modifiche post-approvazione per prodotti non coperti da altre linee guida o dagli standard pertinenti della Farmacopea. Per quanto riguarda la terapia in vitro (IVRT) e la terapia in vitro (IVPT), gli aspetti più rilevanti delle linee guida sono:
Sviluppo farmaceutico
-
Sviluppo della formulazione
Lo sviluppo della formulazione deve essere allineato al QTTP (Quality Target Product Profile), con test adeguati per caratterizzare e controllare i CQA (Critical Quality Attributes) come la facilità di somministrazione, la durata d’uso e le prestazioni del prodotto come la dissoluzione, l’IVRT e, se appropriato, l’IVPT.
-
Caratterizzazione del prodotto
La caratterizzazione è necessaria per facilitare la gestione del ciclo di vita e, se necessario, l’equivalenza del prodotto. I test di prestazione chiave dovrebbero includere la dissoluzione delle sospensioni, l’IVRT e l’IVPT, se necessario. Le prestazioni del prodotto devono essere dimostrate stabili durante la conservazione.
Strategia di controllo
I CQA cruciali per il controllo del rilascio del farmaco devono essere gestiti con attenzione, con test come IVRT e IVPT (se pertinenti). Altri parametri (ad esempio microscopia, DSC, reologia) possono essere utilizzati se si dimostrano più discriminanti nel controllo del rilascio del farmaco. Inoltre, eventuali limiti dei test di prestazione (ad esempio, dissoluzione, IVRT) inclusi nelle specifiche devono essere giustificati con riferimento a lotti clinici di comprovata efficacia e sicurezza.
Programma di stabilità
I test di stabilità devono garantire la qualità e l’efficacia del prodotto nel tempo, mentre l’IVRT o altri test di prestazione confermano la costanza della durata di conservazione.
Raccomandazioni di equivalenza per prodotti topici
Questa parte delle linee guida si applica ai nuovi prodotti cutanei che vogliono dimostrare l’equivalenza terapeutica con un medicinale esistente. È applicabile anche alle modifiche post-approvazione quando si prevede un potenziale impatto sulla qualità, sicurezza o efficacia a seguito di una valutazione del rischio.
La guida afferma inoltre specificamente che “nel caso di domande che si basano sulla letteratura per dimostrare la sicurezza e l’efficacia del medicinale, la pertinenza della letteratura dovrebbe essere supportata da dati di collegamento di equivalenza al prodotto descritto nella letteratura”.
Come affermato in precedenza, l’EMA raccomanda un approccio graduale per dimostrare l’equivalenza. Questo consente alle aziende farmaceutiche di sapere in anticipo quali test eseguire sui loro prodotti (formulazioni semplici come soluzioni o gel, o formulazioni complesse come emulsioni), con l’obiettivo principale di evitare studi clinici completi.
Nel decidere quale approccio e albero decisionale selezionare, l’EMA prende in considerazione la composizione qualitativa, la composizione quantitativa e la caratterizzazione fisico-chimica e strutturale dei prodotti cutanei:
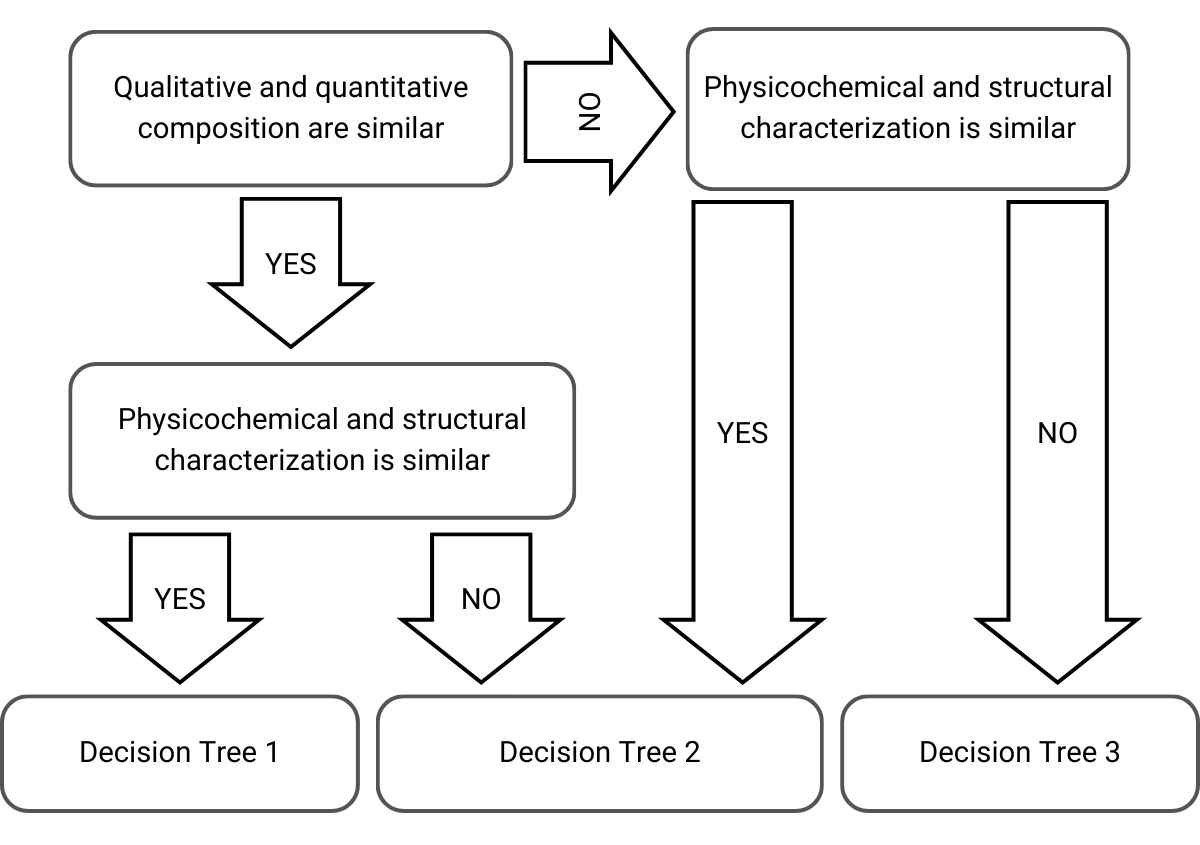
Figura 1) Selezione dell’albero decisionale nell’approccio graduale adattato dalle linee guida dell’EMA
Quindi, a seconda di queste considerazioni, l’approccio graduale è il seguente per i successivi alberi decisionali che portano a ponti accettabili o rifiuti:
Albero decisionale 1: Stessa composizione qualitativa e quantitativa e stessa caratterizzazione fisico-chimica e strutturale
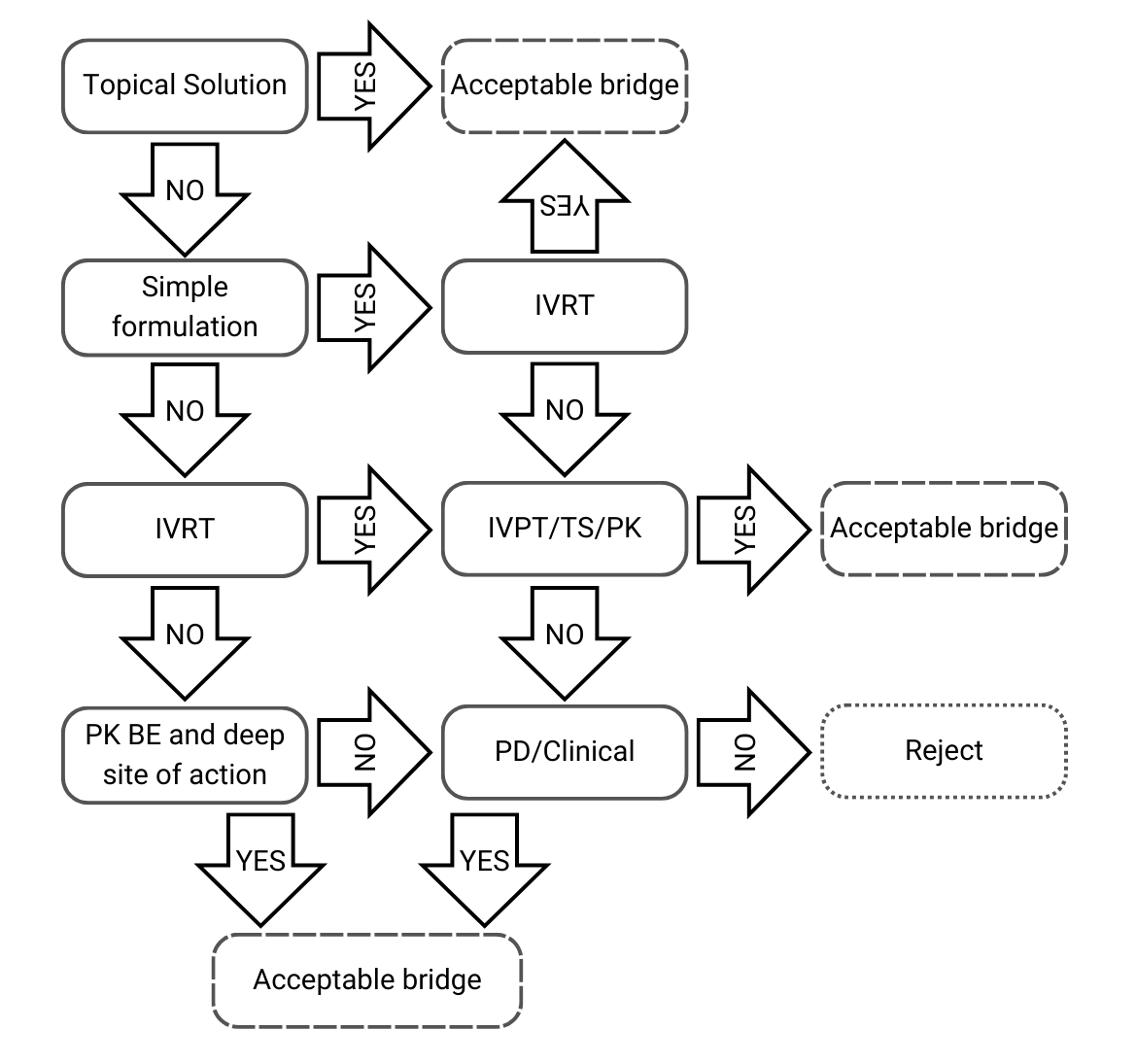
Figura 2) Albero decisionale 1 dalle linee guida dell’EMA
Albero decisionale 2: Piccole differenze nelle composizioni qualitative e quantitative e/o nella caratterizzazione fisico-chimica e strutturale
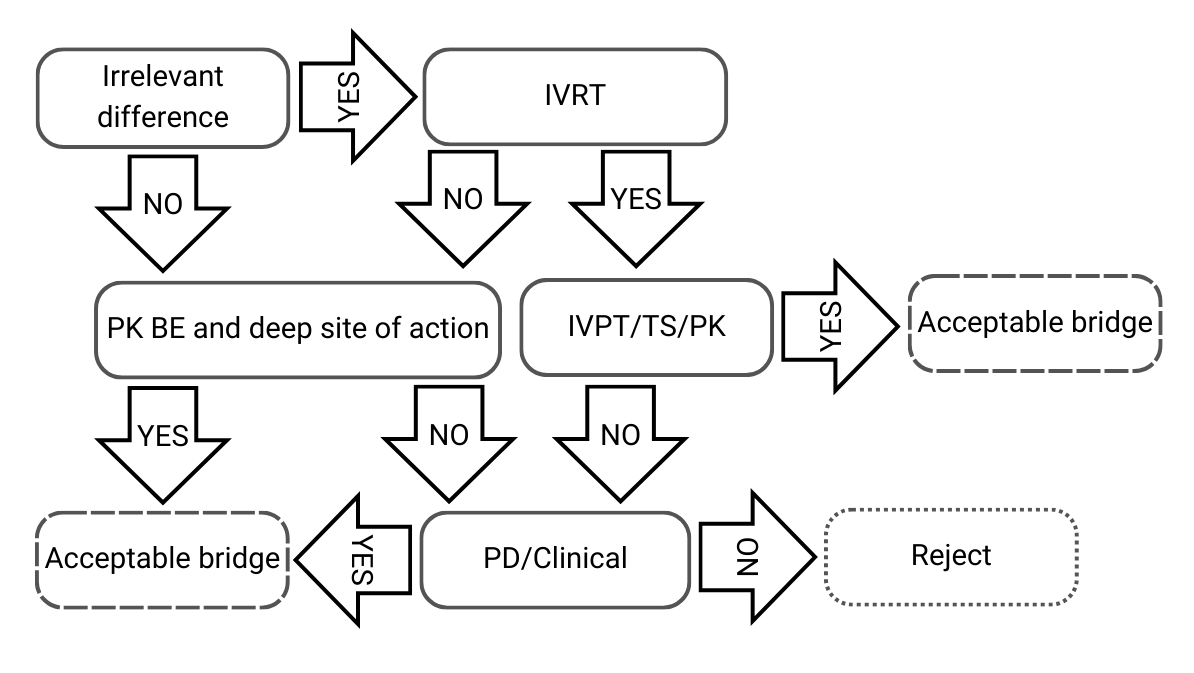
Figura 3) Albero decisionale 2 dalle linee guida dell’EMA
Albero decisionale 3: Diverse composizioni qualitative e quantitative e diverse caratterizzazioni fisico-chimiche e strutturali
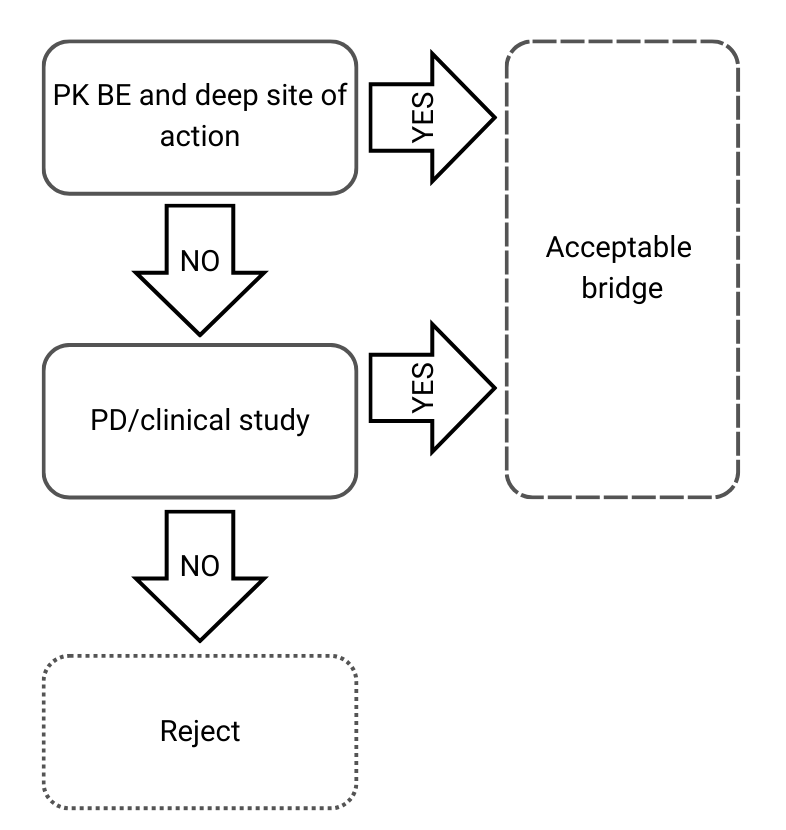
Figura 4) Albero decisionale 3 dalle linee guida dell’EMA
Come si evince da questi alberi decisionali, a seconda delle somiglianze tra i prodotti, l’EMA propone diversi approcci graduali che i produttori dovrebbero prendere in considerazione.
L’esperienza di Kymos Group con gli studi IVRT e IVPT
Kymos Group dispone di un team specializzato che si occupa di rilascio di farmaci e tecniche per prodotti topici dal 2017 e vanta una vasta esperienza negli studi IVRT e IVPT. I nostri metodi sono stati sviluppati in conformità con la bozza di queste linee guida del 2018, e i nostri scienziati hanno già familiarità con la versione definitiva.
Abbiamo suddiviso il nostro catalogo di servizi di rilascio di farmaci e di assorbimento percutaneo in due gruppi principali:
- IVRT: Misuriamo le quantità e le velocità di rilascio del farmaco utilizzando membrane artificiali per sviluppare e convalidare metodi per diverse formulazioni. Questi metodi possono essere utilizzati in studi comparativi per valutare l’equivalenza e anche per eseguire il controllo di qualità dei lotti di produzione.
- IVPT: Misuriamo le quantità permeate transdermicamente, le velocità di flusso e la distribuzione degli strati su campioni di pelle per studi di bioequivalenza. Possiamo anche contribuire all’ottimizzazione e al confronto delle formulazioni con studi di assorbimento percutaneo.
Il nostro laboratorio è una delle poche strutture europee a offrire test di assorbimento percutaneo certificati GLP e GMP per prodotti cutanei con i più recenti strumenti automatizzati Hanson a diffusione verticale Phoenix (test delle cellule di Franz).
Grazie alla profonda conoscenza delle nuove linee guida, il nostro team scientifico è pronto a supportare i clienti dallo sviluppo della formulazione alla presentazione normativa di nuove domande di autorizzazione all’immissione in commercio, ma anche per i prodotti cutanei che desiderano dimostrare l’equivalenza con i medicinali esistenti, seguendo l’approccio graduale dell’EMA.
Conclusioni
L’adozione delle nuove linee guida EMA sulla qualità e l’equivalenza dei prodotti topici rappresenta un importante passo avanti verso un approccio più armonizzato, ma anche più strutturato, all’analisi di queste tipologie di medicinali. Le sue raccomandazioni graduali per dimostrare l’equivalenza utilizzando tecniche come la radioterapia in vitro (IVRT) e la radioterapia in vitro (IVPT) semplificano il processo di immissione sul mercato di nuovi farmaci generici, evitando la necessità di studi clinici costosi e dispendiosi in termini di tempo.
In qualità di CRO leader nei settori IVRT e IVPT, Kymos Group e il suo team sono pronti a supportare clienti e partner in ogni fase, garantendo un percorso agevole verso l’approvazione per la commercializzazione in Europa, nel rispetto delle più recenti linee guida.
Per maggiori informazioni sulle linee guida EMA o per assistenza con i vostri progetti di assorbimento percutaneo, contattate commercial@kymos.com . Saremo lieti di fornirvi consulenza e supporto dettagliati.
