
Die Europäische Arzneimittel-Agentur (EMA) gab im Oktober 2024 die Verabschiedung der lang erwarteten Leitlinie zur Qualität und Äquivalenz topischer Arzneimittel bekannt. Das endgültige Dokument wurde bereits 2018 entworfen und tritt im April 2025 unter dem offiziellen Titel „ Leitlinie zur Qualität und Äquivalenz lokal angewendeter, lokal wirkender Hautarzneimittel “ in Kraft.
Unsere Abteilungen für IVRT & IVPT sowie Qualitätssicherung haben die wichtigsten Aspekte der EMA-Leitlinie zu IVRT und IVPT zusammengefasst und in Qualitätsempfehlungen und den Abschnitt zur therapeutischen Äquivalenz unterteilt. Abschließend haben sie die Erfahrungen der Kymos-Gruppe mit der Umsetzung der Leitlinie seit deren vorherigem Entwurf im Jahr 2018 dargelegt.
Wie lautet die neue EMA-Leitlinie für topische Produkte für 2024?
Wie der Titel bereits andeutet, bezieht sich diese Leitlinie auf lokal angewendete und lokal wirkende Arzneimittel zur Anwendung auf der Haut, kann aber auch für andere Produkte wie Präparate zur Anwendung am Ohr oder Auge sowie für lokal wirkende Vaginalpräparate relevant sein. Diese neue Verordnung ist in Qualitäts- und Äquivalenzempfehlungen unterteilt und führt einen strukturierteren Ansatz zur Bewertung von Hautpräparaten ein.
Der zweite Teil bezüglich der Äquivalenz gilt nicht für biologische Arzneimittel, pflanzliche Arzneimittel, Arzneimittel, deren Wirksamkeitsäquivalenz in klinischen Studien nachgewiesen wurde, und Arzneimittel, bei denen die Darreichungsform des Prüf- und des Referenzpräparats nicht identisch ist. Dieser Ausschluss ist insbesondere bei systemisch wirkenden Darreichungsformen wie transdermalen Pflastern von Bedeutung.
Mit diesen jüngsten Aktualisierungen betont die EMA einen strukturierteren und schrittweisen Ansatz, der den Prozess des Nachweises der therapeutischen Äquivalenz vereinfachen soll. Im Fokus stehen dabei In-vitro-Methoden (In-vitro-Freisetzungstest oder IVRT und In-vitro-Permeationstest oder IVPT) und pharmakokinetische (PK) Methoden, die kostensparende und zeiteffiziente Alternativen zu klinischen Studien darstellen.
Qualitätsempfehlungen für topische Produkte
Die Qualitätsempfehlungen der Leitlinie gelten für Neuzulassungsanträge und Änderungen nach der Zulassung von Produkten, die nicht durch andere Leitlinien oder die einschlägigen Arzneibuchstandards abgedeckt sind. Bezüglich IVRT und IVPT sind die wichtigsten Aspekte der Leitlinie:
Pharmazeutische Entwicklung
-
Formulierungsentwicklung
Die Formulierungsentwicklung sollte auf das QTTP (Quality Target Product Profile) abgestimmt sein, mit geeigneten Tests zur Charakterisierung und Kontrolle von CQAs (Critical Quality Attributes) wie z. B. Anwendungsfreundlichkeit, Anwendungsdauer und Produktleistung wie Auflösung, IVRT und gegebenenfalls IVPT.
-
Produktcharakterisierung
Die Charakterisierung ist für das Lebenszyklusmanagement und gegebenenfalls für die Produktäquivalenz erforderlich. Zu den wichtigsten Leistungstests sollten die Auflösung von Suspensionen, IVRT und gegebenenfalls IVPT gehören. Die Produktleistung muss während der Lagerung stabil sein.
Kontrollstrategie
Kritische Qualitätsmerkmale (CQAs), die für die Kontrolle der Wirkstofffreisetzung entscheidend sind, sollten sorgfältig überwacht werden, gegebenenfalls mithilfe von Tests wie IVRT und IVPT. Weitere Parameter (z. B. Mikroskopie, DSC, Rheologie) können herangezogen werden, wenn sie sich bei der Kontrolle der Wirkstofffreisetzung als aussagekräftiger erweisen. Darüber hinaus müssen alle in der Spezifikation festgelegten Grenzwerte für Leistungstests (z. B. Auflösung, IVRT) durch klinische Chargen mit nachgewiesener Wirksamkeit und Sicherheit begründet werden.
Stabilitätsprogramm
Stabilitätsprüfungen müssen die Qualität und Wirksamkeit des Produkts über die Zeit gewährleisten, wobei IVRT oder andere Leistungstests die Beständigkeit über die Haltbarkeitsdauer bestätigen.
Empfehlungen zur Gleichwertigkeit von topischen Produkten
Dieser Teil der Leitlinie gilt für neue Hautarzneimittel, die die therapeutische Äquivalenz mit einem bereits zugelassenen Arzneimittel nachweisen wollen. Er ist auch auf Änderungen nach der Zulassung anwendbar, wenn nach einer Risikobewertung potenzielle Auswirkungen auf Qualität, Sicherheit oder Wirksamkeit zu erwarten sind.
Der Leitfaden stellt außerdem ausdrücklich fest, dass „bei Anträgen, die sich auf Literatur stützen, um die Sicherheit und Wirksamkeit des Arzneimittels nachzuweisen, die Relevanz der Literatur durch Äquivalenzdaten zu dem in der Literatur beschriebenen Produkt untermauert werden sollte“.
Wie bereits erwähnt, empfiehlt die EMA ein stufenweises Vorgehen zum Nachweis der Äquivalenz. Dadurch wissen Pharmahersteller im Voraus, welche Tests sie für ihre Produkte durchführen müssen (einfache Formulierungen wie Lösungen oder Gele oder komplexe Formulierungen wie Emulsionen), und das Hauptziel ist es, vollständige klinische Studien zu vermeiden.
Bei der Entscheidung über die Vorgehensweise und den Entscheidungsbaum berücksichtigt die EMA die qualitative und quantitative Zusammensetzung sowie die physikalisch-chemischen und strukturellen Eigenschaften der Hautprodukte:
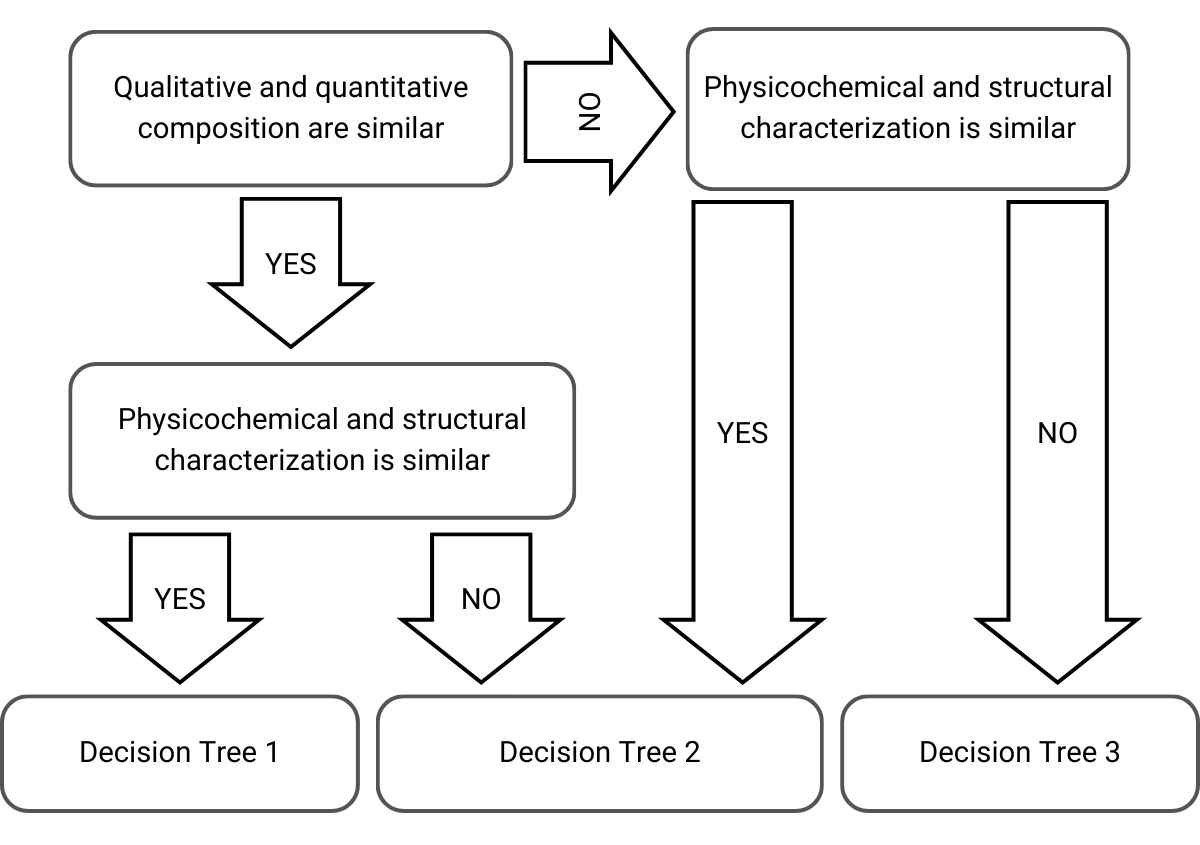
Abbildung 1) Auswahl des Entscheidungsbaums im schrittweisen Verfahren, adaptiert nach der EMA-Richtlinie
Abhängig von diesen Überlegungen ergibt sich dann folgendes schrittweises Vorgehen bei den nächsten Entscheidungsbäumen, die zu akzeptablen Brücken oder Ablehnungen führen:
Entscheidungsbaum 1: Gleiche qualitative und quantitative Zusammensetzung sowie gleiche physikalisch-chemische und strukturelle Eigenschaften
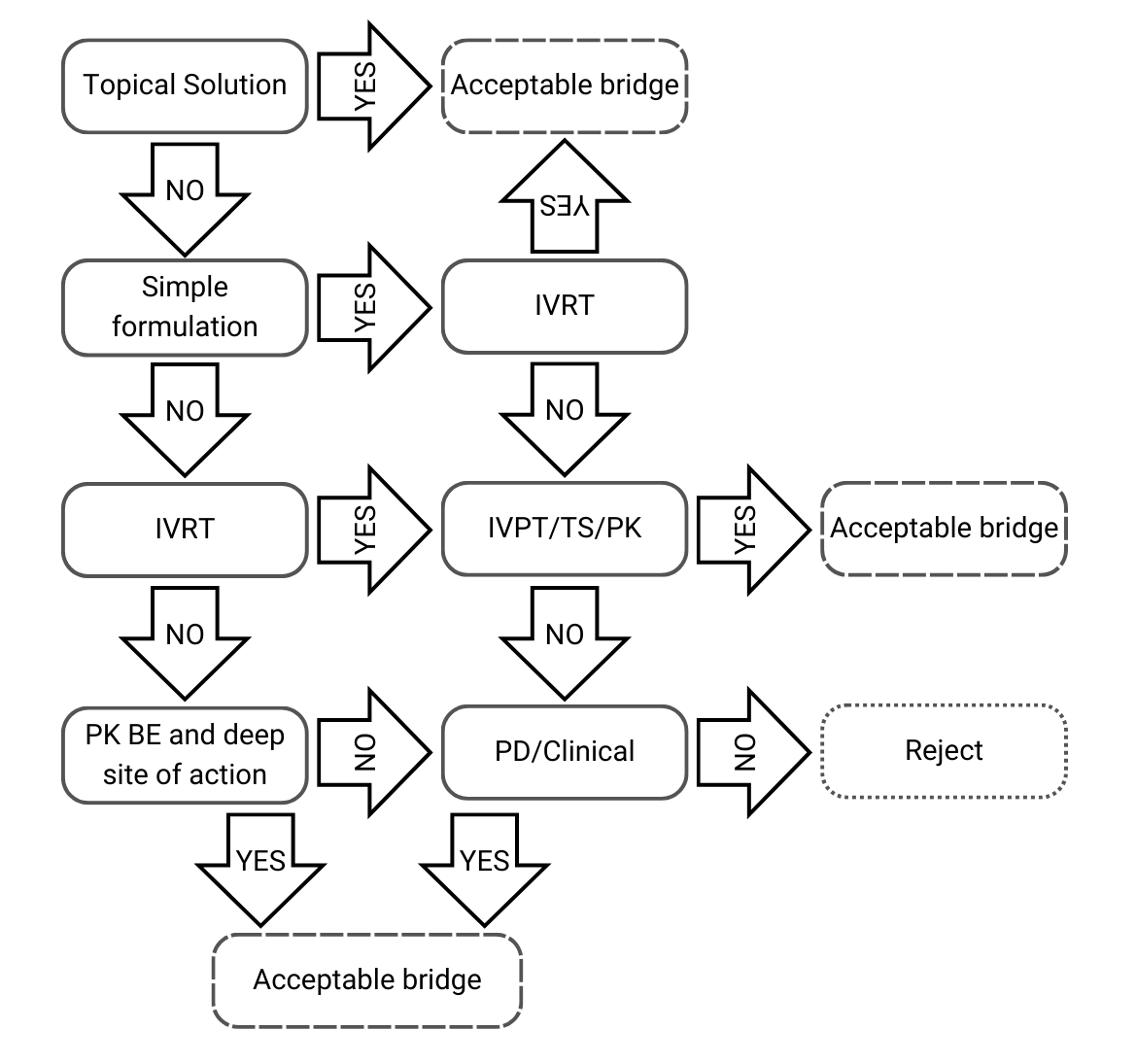
Abbildung 2) Entscheidungsbaum 1 aus der EMA-Leitlinie
Entscheidungsbaum 2: Geringfügige Unterschiede in der qualitativen und quantitativen Zusammensetzung und/oder der physikalisch-chemischen und strukturellen Charakterisierung
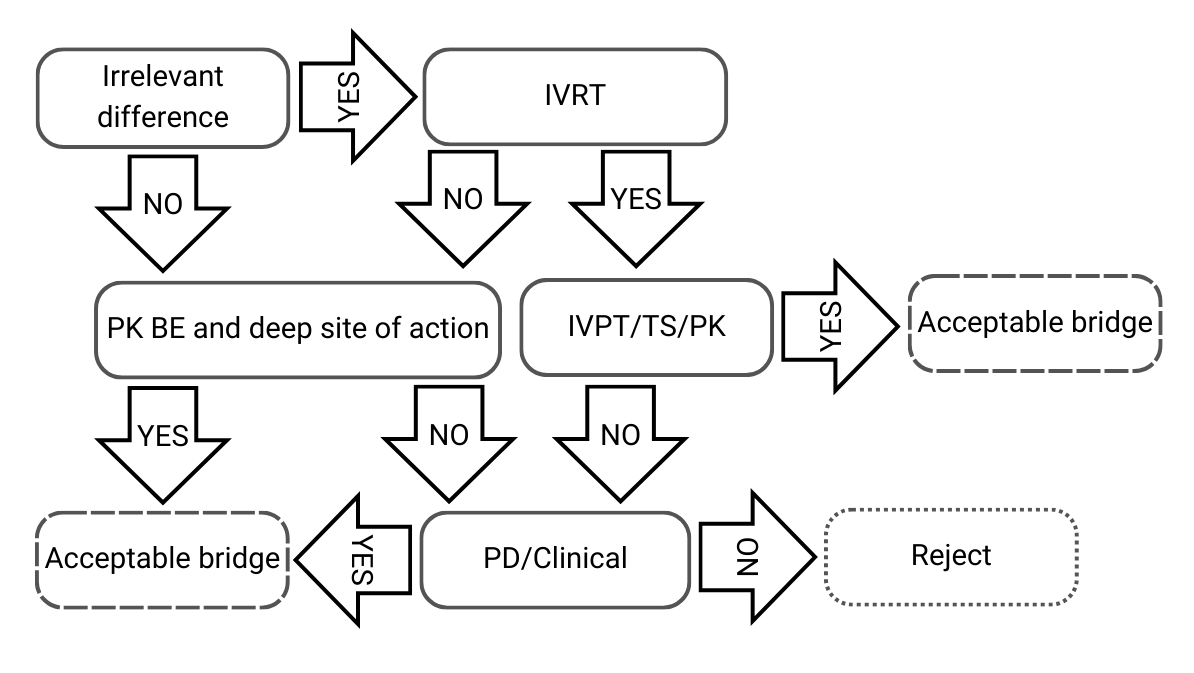
Abbildung 3) Entscheidungsbaum 2 aus der EMA-Leitlinie
Entscheidungsbaum 3: Unterschiedliche qualitative und quantitative Zusammensetzungen sowie unterschiedliche physikalisch-chemische und strukturelle Charakterisierung
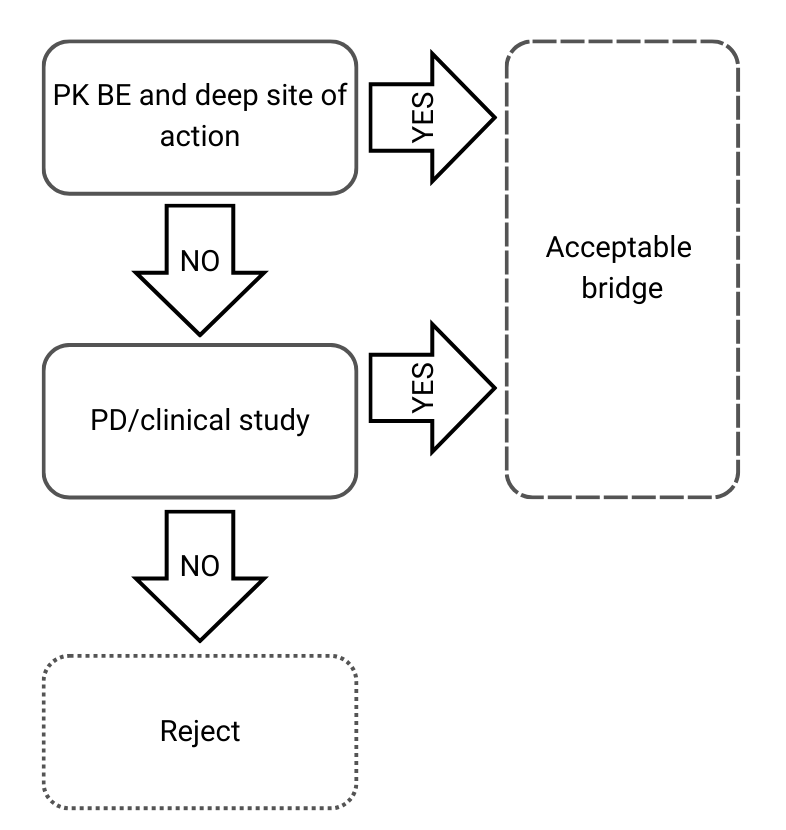
Abbildung 4) Entscheidungsbaum 3 aus der EMA-Leitlinie
Wie aus diesen Entscheidungsbäumen ersichtlich, bietet die EMA je nach Ähnlichkeit der Produkte verschiedene schrittweise Vorgehensweisen an, die Hersteller in Betracht ziehen sollten.
Die Erfahrungen der Kymos Group mit IVRT- und IVPT-Studien
Die Kymos Group verfügt seit 2017 über ein spezialisiertes Team, das sich mit Wirkstofffreisetzungstechniken für topische Produkte befasst und umfangreiche Erfahrung mit IVRT- und IVPT-Studien besitzt. Unsere Methoden wurden in Übereinstimmung mit dem Entwurf dieser Leitlinie aus dem Jahr 2018 entwickelt, und unsere Wissenschaftler sind bereits mit der finalen Fassung vertraut.
Wir haben unser Angebot an Dienstleistungen zur Wirkstofffreisetzung und perkutanen Absorption in zwei Hauptgruppen unterteilt:
- IVRT: Wir messen Wirkstofffreisetzungsmengen und -raten mithilfe künstlicher Membranen, um Methoden für verschiedene Formulierungen zu entwickeln und zu validieren. Diese Methoden können in Vergleichsstudien zur Bewertung der Äquivalenz sowie zur Qualitätskontrolle von Produktionschargen eingesetzt werden.
- IVPT: Wir messen transdermal permeierte Mengen, Flussraten und Schichtverteilung anhand von Hautproben für Bioäquivalenzstudien. Darüber hinaus unterstützen wir die Optimierung und den Vergleich von Formulierungen durch Studien zur perkutanen Absorption.
Unser Labor ist eine der wenigen europäischen Einrichtungen, die sowohl GLP- als auch GMP-zertifizierte perkutane Absorptionsassays für Hautprodukte mit den neuesten automatisierten Hanson Phoenix-Instrumenten zur vertikalen Diffusion (Franz-Zellen-Test) anbieten.
Mit einem tiefen Verständnis der neuen Richtlinien ist unser wissenschaftliches Team bereit, Kunden von der Formulierungsentwicklung bis zur behördlichen Einreichung neuer Zulassungsanträge zu unterstützen, aber auch für Hautprodukte, die die Gleichwertigkeit mit bestehenden Arzneimitteln nach dem stufenweisen Ansatz der EMA nachweisen wollen.
Schlussfolgerungen
Die Übernahme der neuen EMA-Leitlinie zur Qualität und Äquivalenz topischer Arzneimittel stellt einen wichtigen Fortschritt hin zu einem harmonisierteren und strukturierteren Ansatz für die Analyse dieser Arzneimittel dar. Ihre schrittweisen Empfehlungen zum Nachweis der Äquivalenz mithilfe von Verfahren wie IVRT und IVPT beschleunigen die Markteinführung neuer Generika und machen aufwändige und zeitintensive klinische Studien überflüssig.
Als führendes CRO im Bereich IVRT und IVPT steht die Kymos Group mit ihrem Team ihren Kunden und Partnern bei jedem Schritt zur Seite und gewährleistet einen reibungslosen Weg zur Marktzulassung in Europa gemäß den neuesten Richtlinien.
Benötigen Sie weitere Informationen zur EMA-Richtlinie oder Unterstützung bei Ihren Projekten zur perkutanen Absorption? Dann kontaktieren Sie uns bitte unter commercial@kymos.com . Wir beraten und unterstützen Sie gerne ausführlich.
